Mostrar más estudios...
0
Salud por estilo de vida
Enfermedades crónicas degenerativas

Laboratorio
Gabinete
Imagenología
Check ups

Equipo Inmunoquímica

Equipo Inmunoquímica
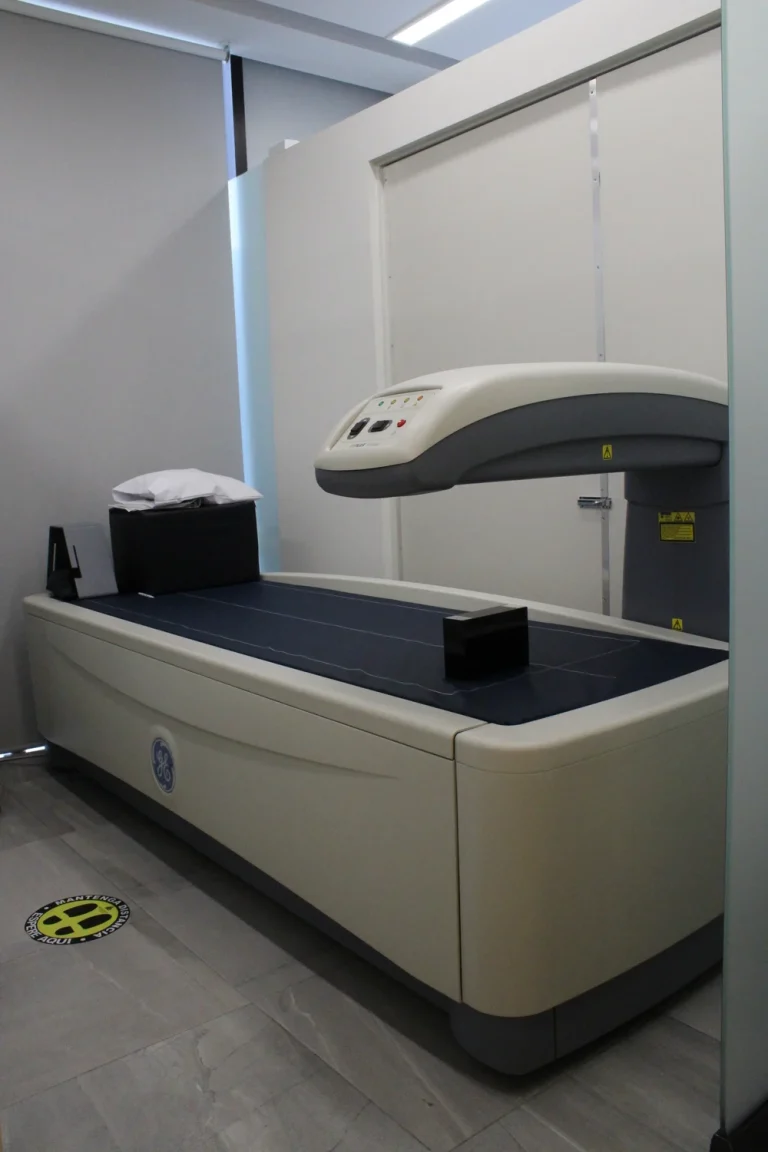
Equipo Densitometría
Equipo Electrocardiograma

Equipo Rayos X
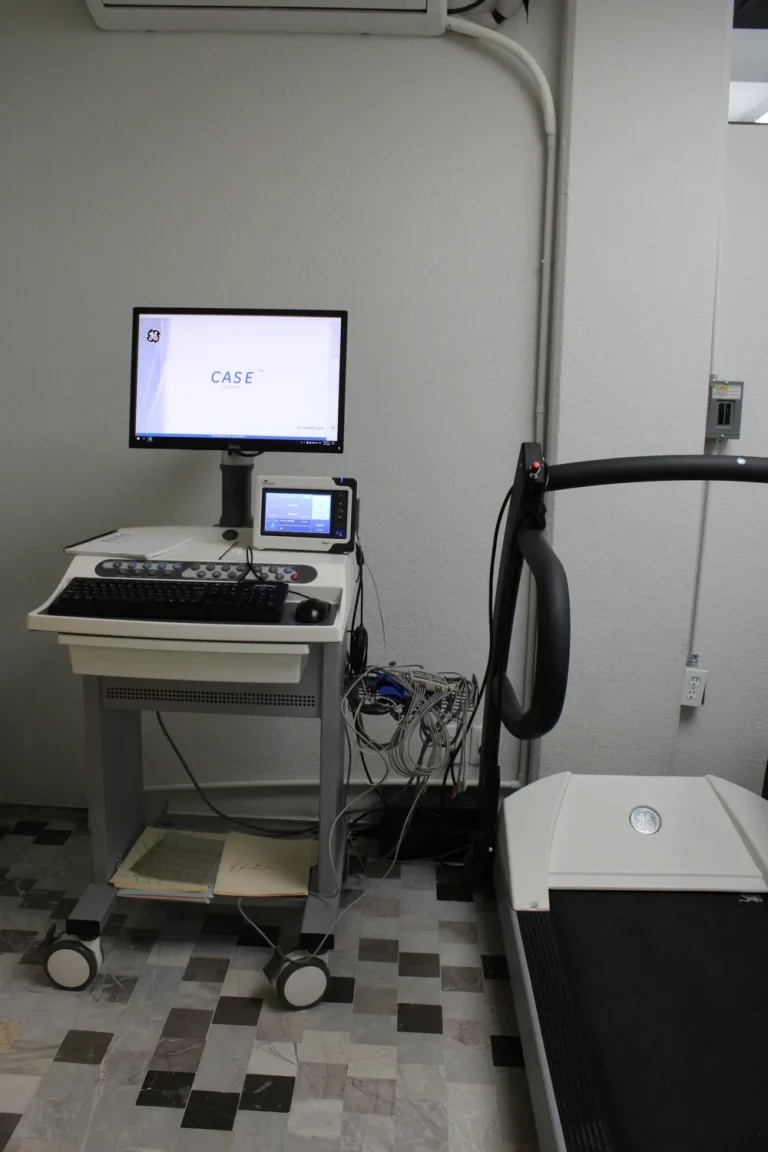
Equipo Prueba de esfuerzo
Equipo Ultrasonido